


At the occasion of the Alphy/AIEM 2023 conference we made a first (beta) Evoluthon contest.
The rules and context can be found below : in one word, one had to reconstruct a tree based on 40 sequences generated by Aevol.
Now that this contest is over, we can share the original tree, to enable anyone to train themselves on this first example ; we will make a bigger contest with new sets of sequences starting in September.
This new contest should provide different level of difficulty and sequences having evolved in different conditions (bottleneck, changing mutation rate…) between sets.
Beta contest : sequences, solution(s), rules
Sequences
Click on the links below to download the sequences in multi-fasta format.
Solution(s)
On the links below you can download :
First the original tree in newick format (in .txt here).
Secondly, if you want to try and discover the ancestor sequence, the true one is on the second link.
Finally, if you want to try and discover the parameters for this simulation (intitial population size, maximum indel size, point mutation rate, small deletion rate, small insertion rate, duplication rate, translocation rate, insertion rate, deletion rate), those are on the last link.
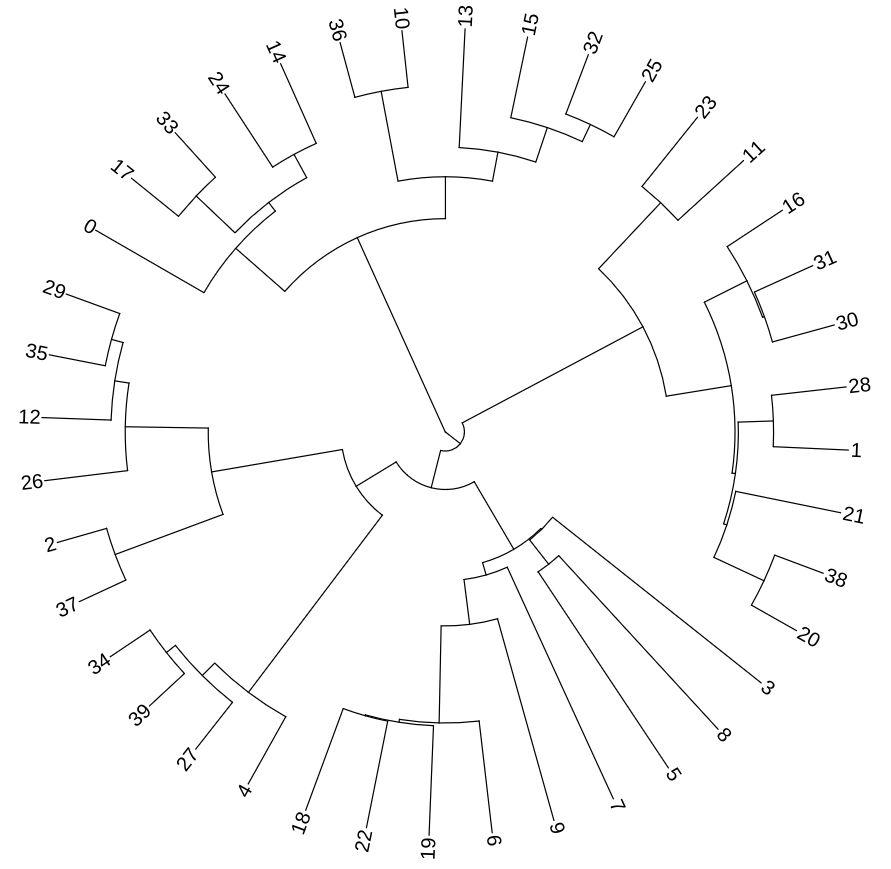
Rules
For this contest, we provide you with 40 DNA sequences of ~40 000 base pairs simulated using Aevol.
These sequences are inspired by bacterial genomes. Each sequence comes from a different species, those different species having evolved in silico starting from one unique genome.
We challenge you to use your favorite tools and methods to reconstruct the closest to the original phylogenetic tree.
You can submit your trees by sending them to hugo.daudey@univ-lyon1.fr using the format of your choosing (newick, NEXUS, phyloxml…).
Also, feel free to add any information you think you found out about this data set (population sizes, selection pressures, notable events, …).
We can then confirm or infirm to you your deductions based on the record we have of the simulation that yielded the sequences.
Context
Molecular evolution methods face a validation issue : we cannot go back in time and check hypotheses or predictions, which concern events that occurred possibly billions of years ago.
Across the whole of scientific literature, the most popular validation process are computer simulations.
Genomic evolution can be simulated in silico over a far greater number of generations than using experimental evolution, and in a less costly way.
New methods are almost always tested on ad hoc simulations, which means simulations created on purpose to test these new methods specifically.
This process inevitably leads to essential features of the tested method being incorporated within the simulations, which implies that those simulations may only serve to test said new methods, and will generate instances failing to represent real data complexity.
Evoluthon is a project that aims at challenging methods with data created by simulations not influenced by said methods to be tested.
To achieve this, we propose Aevol as an impartial benchmarking tool.
Aevol is an open-source digital genetics platform that captures the evolutionary process using genetic algorithms and individual based modeling.
Digital organisms in Aevol reproduce, compete and mutate, evolving for hundreds of thousands of generations under typical Darwinian dynamics.
Hugo Daudey (1), Marco Foley (2), Jonathan Rouzaud-Cornabas (2)(3), Vincent Daubin (1), Bastien Bousseau (1), Éric Tannier (1)(2), Guillaume Beslon (2)(3)
(1) Université Claude Bernard Lyon 1, CNRS, Laboratoire de Biométrie et Biologie Evolutive, Villeurbanne, France
(2) Centre de recherche Inria de Lyon (Équipe BEAGLE), Villeurbanne, France
(3) INSA-Lyon, Villeurbanne, France

Frequently asked questions:
Why Aevol? It is necessary that the simulation software has been developed by a team who doesn’t develop inference methods. Otherwise the simulation will be partial: oriented towards particular models. Aevol is the only one we know in this case, producing sequences usable by bioinformatics methods.
What are the genomes supposed to look like? They are simulated with a darwinian process of mutation/selection, and are constrained by some limits in available computing time. Absence of sex, and their size make them compare to bacterial genomes. Many features however may not look like real bacterial genomes. Imagine them as extra-terrestrial life and see what you can deduce from their evolution.